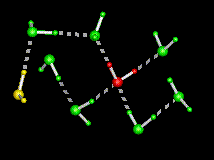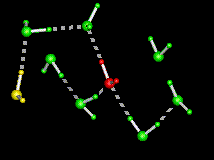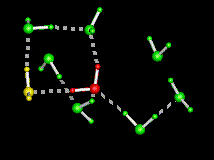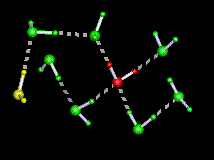
| This figure shows the computed minimum energy path of a water molecule rotating in ice, i.e. as it interchanges its two hydrogens. Initially, the rotating water at the centre (in red) is hydrogen bonded to its four immediate tetrahedral neighbours (in green), while the water on the left (in yellow) is termed a L-defect because there is no hydrogen between it and the central water. During the rotation, note the formation of an intermediate hydrogen bond between the central water and the L-defect, followed by a bifurcated hydrogen bond to one of the water molecules in green. Both contribute to decreasing the energy along the path. The part of the path where the central water makes only three hydrogen bonds (the "free" hydrogen faces the viewer) is the transition state, which has the highest energy. The maximum displacement of the central water is 1.5A. | |

The above figure shows the sequence of motions of the rotating water, shown along the reaction coordinate (lambda). The arrows show the rotation axes. The first rotation is orthogonal to the plane of the image across the O-H2 bond while the second rotation is in the plane of the image across an axis passing through O and orthogonal to the plane of the image. The initial and final states are depicted at the ends of the figure (note the resulting interchange of the two hydrogens).
The process of water flip is similar in both
ice
and the protein and is a complex motion, involving two successive
rotations
around mutually orthogonal axes. The process is governed by short range
interactions. The ability of the water to flip is enhanced by the
presence
of alternative hydrogen bonding sites or "defects" in its immediate
neighbourhood
which help break the tetrahedral symmetry around the oxygen. The motion
was also found to be characterized by one unique path.
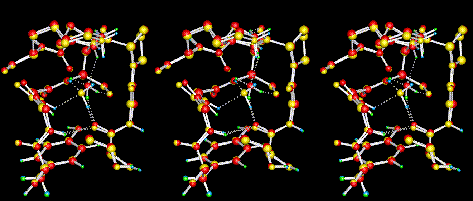
In the next stereo figure (wall/cross-eyed),
the ground state (red heavy atoms andgreen
hydrogens) and transition state (yellow
heavy atoms and blue
hydrogens) structures of the water-flip in
BPTI are shown.
Rate of rotation in BPTI:
The computation of the transition state enabled
the calculation of the vibrational density of states for both the
ground
and the transition states (in fact this is the first time that a
complete
normal mode analysis of a large system as a protein in its transition
state
has been computed). Vibrational frequencies up to 620 cm-1
were
found to contribute to the vibrational entropy of activation, which was
computed to be 8.6 cal/mol/K. This leads to a lowering of the free
energy
of the isomerisation by
2.58 kcal/mol at 300K and leads to
an estimate of a lower bound for the rotational correlation time of
45ns
(the
computed free energy of activation is 6.08 kcal/mol).
Go to Home of S. Fischer